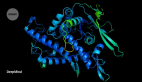
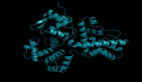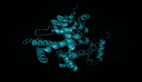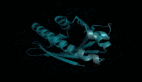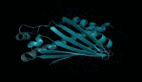
提到AI4Science, 尤其是AI在生物学领域的应用,DeepMind的AlphaFold已经成为了「出圈」的代表作,是生物学和计算交叉融合的里程碑。
2020年底,第一代AlphaFold就已经能以原子级别的精度预测蛋白质的三维形状。然而,这仅仅是一个开始。
一个不那么明显的事实是,与Meta FAIR研发的ESM系列不同,AlphaFold并非不是使用LLM构建的,而是依赖于一种名为「多序列比对」(MSA)的方法,是生物信息学领域一种较为传统的做法。
当全球的目光聚焦于AlphaFold时,大语言模型在生物学领域的潜力或许被低估了。
去年,AI专栏作家、Radical VC合伙人Rob Toews在《福布斯》杂志上发表了一篇文章,预言LLM在生物领域的潜力。
 图片
图片
相比速度慢且计算量大的MSA方法,LLM在规模化预测方面有更大的优势,而且能产生同样准确,甚至更准确的结构预测。
此外,语言模型能够对蛋白质的潜在特征空间产生更普遍的理解,这为蛋白质科学开辟了令人兴奋的可能性。、
无独有偶,去年11月,微软研究院的AI4Science部门和Azure量子计算部门发表了一篇长达230页的报告,其中着重强调了GPT-4协助生物学领域前沿研究的巨大潜力。
 图片
图片
论文地址:https://arxiv.org/pdf/2311.07361
虽然只在人类语言上进行训练,但GPT-4依旧表现出了对「蛋白质语言」的理解和推理能力。如图2所示,根据对应的DNA序列,模型可以正确预测MYC蛋白上转录因子的结合位点。
 图片
图片
最近,Nature也收录了一篇GPT-4与生物学进行结合的研究,用更详实的实验和数据佐证了微软这篇报告的结论。
研究人员发现,GPT-4能以出人意料的精度对氨基酸、多肽和蛋白质结构进行建模。
 图片
图片
论文地址:https://www.nature.com/articles/s41598-024-69021-2
虽然相比AlphaFold 3,GPT-4的建模能力只算是初阶,实际应用有限,但考虑到GPT-4的目标是建模人类自然语言,并不是专门为结构生物学任务而开发的,因此达到这种精度才令人惊讶。
作者指出,他们尚不清楚GPT-4这种「触类旁通」的具体机制,需要更广泛的研究才能得出明确结论。但这篇论文的结果无疑揭示了生成模型新的潜力和探索方向。
「无师自通」,GPT-4高精度建模
单个氨基酸结构的建模
氨基酸是蛋白质的最小组成单位,其原子组成和几何参数已经得到了很好的表征,因此非常适合基本结构建模任务。
实验通过prompt给出最少的必要上下文信息,让GPT-4以PDB格式对20个标准氨基酸进行建模,同时还纳入了GPT-3.5作为性能对比。
 图片
图片
对每个单独的氨基酸,研究人员都用相同的prompt输入5次迭代运行以监控一致性。
结果发现,模型可以生成含有主链和侧链原子坐标值的氨基酸3D结构,PDB格式渲染后的可视化效果如下图所示:
 图片
图片
图d、e展示了GPT-4生成主链键长和角度的准确性(蓝色),虽然准确度各不相同,但都聚集在实验确定的参考值(红色)附近。
侧链的预测结果也有极高的精度,近90%的键长差值在0.1 Å以内,近80%的键角误差在10°以内,而且在所有指标上,GPT-4相比GPT-3.5的预测准确性都有较大幅度的提升。

α-螺旋结构的建模
α-螺旋是蛋白质中最常见且被广泛研究的二级结构。研究人员表示,虽然使用各种prompt进行了多次尝试,GPT-4和GPT-3.5都无法像模拟氨基酸一样准确地生成α-螺旋多肽链的结构。
有趣的一点是,GPT-4习惯于用数学公式描述α-螺旋的空间结构参数:
 图片
图片
由于单独使用GPT-4效果不佳,实验人员尝试整合Wolfram插件,这是由Wolfram-Alpha开发的适配GPT-4模型的数学计算扩展。
 调用Wolfram插件并得到响应
调用Wolfram插件并得到响应
和之前的实验一样,给GPT-4输入上下文prompt,对含有10个氨基酸的多肽进行PDB格式的结构预测。
其中,多肽上的每个位点都单独进行一轮迭代,每个多肽同样重复预测5轮。
 实验流程示意图
实验流程示意图
每次迭代中,实验人员还会进行最多3次尝试,通过prompt给模型提出改进建议,比如提示螺旋的直径过大或过小等等,相当于提供专业人员的校准和监督:
 图片
图片
可以看出,GPT-4和Wolfram结合后,基本可以捕捉到α-螺旋的整体结构
 图片
图片
结构建模的准确度可以用「均方根偏差」(root-mean-square deviation, RMSD)进行量化。
下图e显示,每次输入prompt迭代后,输出的准确性都有明显提升。经过两次基于提示的改进后,近90%的预测结果RMSD误差已经小于0.5Å,但相比AlphaFold 2的结果依旧有一定差距(下图f)。
 图片
图片
结构相互作用分析
实验主要关注抗病毒药物和病毒蛋白之间的结构相互作用的定性分析,以nirmatrelvir-SARS-CoV-2为例。
其中SARS-CoV-2是导致COVID-19的一种病毒株;nirmatrelvir中文名为奈玛特韦,是辉瑞研制的COVID-19口服药帕昔洛韦的主要成分之一。
实验中,给GPT-4提供PDB格式的nirmatrelvir晶体结构,提示模型进行配体检测和相互作用检测。
 配体相互作用分析流程
配体相互作用分析流程
整体的实验过程如下图所示:
 图片
图片
结果发现,GPT-4正确识别出了nirmatrelvir配体,并在输出的PDB文件中用「4WI」标识了出来。
 图片
图片
在此基础上进行的相互作用检测中,GPT-4在结合位点内列出了5种氨基酸,恰好涵盖了4种直接结合nirmatrelvir配体的氨基酸。
此外,模型还描述出了几种可能干扰结合过程的突变,其中大部分是合理的,但有一些突变无关紧要。
值得注意的是,其中提到的GLU A166突变已被证明对nirmatrelvir结合极其有害,且会造成临床治疗中的病毒耐药性。
 图片
图片
 图片
图片
讨论和结论
这篇报告探索性地展示了,在基本结构生物学建模和药物相互作用分析等领域,GPT-4模型的已有能力和局限性。
鉴于生物信息学常用的AI工具,比如AlphaFold、RoseTTAFold 和蛋白质语言模型等,与自然语言领域的LLM存在技术路线上的固有差距,这种发现呈现出独特的新颖性。
虽然GPT-4在标准氨基酸结构建模,以及集成Wolfram后在α-螺旋结构上的建模都有较好的表现,但其中发生的零星错误也不能忽视。即使是引入最小规模的错误,也可能对结构模型和相关的生物学解释非常不利。
实验最后一节中,在预测可能干扰相互作用的蛋白质突变时,GPT-4展现出了非常强大的能力,这对药物发现和开发将会非常有用。
GPT-4为什么能发展出蛋白质的结构建模能力?
一种推测是,训练数据中可能有关于原子坐标的信息,但这不能解释模型输出的结构中含有的几何变异性,而且结构复杂性似乎是预测结果的限制因素。
更有可能的解释是,GPT-4的建模是从头开始运行的,因为响应中包含了原子坐标和数学计算公式,Wolfram插件和prompt给出的改进信息也大大提升了α-螺旋结构建模的准确性,证明预测时发生了推理过程。
参考资料: