












化学反应是药物设计和有机化学研究的基础。研究界越来越需要一种能够有效捕获化学反应基本规则的大规模深度学习框架。
近日,来自北京大学和望石智慧的研究团队提出了一种新方法来弥合基于反应的分子预训练和生成任务之间的差距。
受有机化学机制的启发,研究人员开发了一个新的预训练框架,使其能够将归纳偏差纳入模型中。所提框架在执行具有挑战性的下游任务中取得了最先进的结果。通过掌握化学知识,生成框架克服了当前依赖少量反应模板的分子生成模型的局限性。在大量的实验中,模型生成了高质量的可合成药物样结构。
总的来说,该研究向各种基于反应的应用程序的大规模深度学习框架迈出了重要一步。
该研究以《Bridging the gap between chemical reaction pretraining and conditional molecule generation with a unified model》为题,于 2023 年 12 月 5 日发布在《Nature Machine Intelligence》上。

论文链接:https://www.nature.com/articles/s42256-023-00764-9
深度学习模型已在众多科学研究领域得到应用。预训练框架有助于新任务的无缝集成,从而加快建模过程,特别是对于标记数据有限的场景。
化学反应是药物设计和有机化学研究的基础。目前,数据挖掘研究和应用已经使深度学习模型能够应用于化学反应。基于这些数据,已经有许多数据驱动的研究深入研究化学反应的表征学习。
表征学习是指从数据中自动学习有用的特征,然后将其用于各种下游任务。现有方法忽略了有机化学的基本理论,限制了其性能。
基于化学反应的分子生成
除了反应分类任务之外,基于化学反应的分子生成也是一个重要的应用。早期的研究总是采用基于模板的逐步分子生成策略。
这些基于模板的方法在很大程度上依赖于预定义的构建模块和反应,这缩小了可访问的化学空间。在反应产物预测领域也发现了类似的趋势,其中基于模板的方法不能外推到复杂的反应;这个问题可以通过使用无模板方法来解决。
在基于反应的分子生成任务中,无模板方法也表现出了优于基于模板方法的泛化优势。然而,现有的无模板分子生成方法只能基于预定义的反应物库生成分子。除此之外,对于药物设计中的先导化合物或先导化合物优化阶段,利用化学反应作为编辑工具来修改给定的结构是更有利的。生成的化学库将重点关注可以用更少的反应步骤合成的化学空间的子集。
一个新、全面的化学反应深度学习框架
在此,研究人员提出了一个新的、全面的化学反应深度学习框架,称为 Uni-RXN。旨在解决两个基本任务:自监督表征学习和条件生成建模。

图示:Uni-RXN 的组成和方法。(来源:论文)
与现有方法不同,研究人员提出了一套专门针对化学反应精心设计的自监督任务。这些任务包括反应中心预测、主反应物与子反应物配对以及反应物-产物配对。在对具有挑战性的反应任务的广泛评估中,Uni-RXN 方法超越了最先进的水平,证明了其有效捕获化学反应领域知识的能力。所获得的有希望的结果为广泛的下游应用铺平了道路。
通过有效捕获化学规则,Uni-RXN 非常适合生成任务。与依赖于从预定义反应物库中选择片段的传统方法不同,Uni-RXN 将分子结构作为输入条件并生成相应反应物的表示,同时保持反应内的排列不变性。利用密集向量相似性搜索包的强大功能,Uni-RXN 能够从大型反应物和试剂库中高效检索反应物。随后,采用反应预测模型来生成产物输出。
与仅探索化学空间的有限子集的基于模板的方法相比,Uni-RXN 在生成更广泛的可合成药物样结构方面表现出卓越的性能。这一特点使其特别适合虚拟 library 枚举,并得到全面统计分析和案例研究的支持。
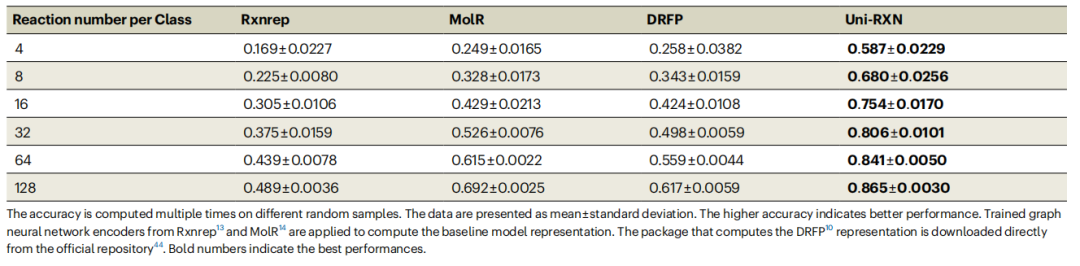
Uni-RXN 方法具有多种优势,能够为具有挑战性的化学反应分类任务派生丰富的表示。Uni-RXN 大幅优于其他基线模型,在每类仅提供 4 个数据点的情况下实现了 58.7% 的准确率。
表 1:化学反应分类的准确度。(来源:论文)
Transformer 模型还可以应用于区分化学反应数据中的优化反应和未优化反应。此外,编码器可以毫不费力地应用于结构条件生成。
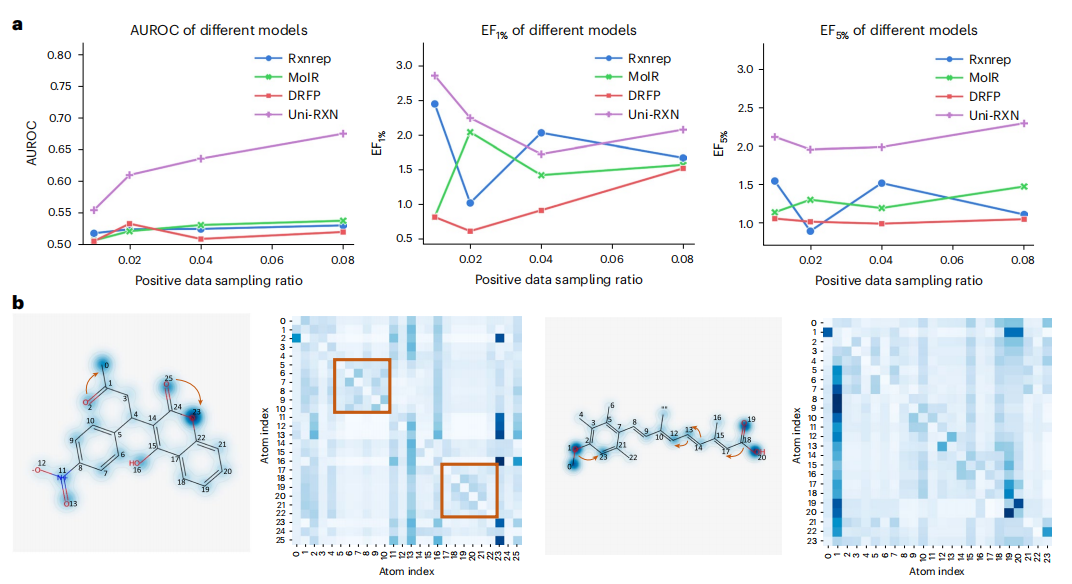
图示:Uni-RXN 的检索性能和注意力权重。(来源:论文)
实验结果强调了所提模型生成的分子的有利特性,使它们非常适合药物发现任务。该模型能够生成具有更多类似药物特性和可合成可及性的分子。
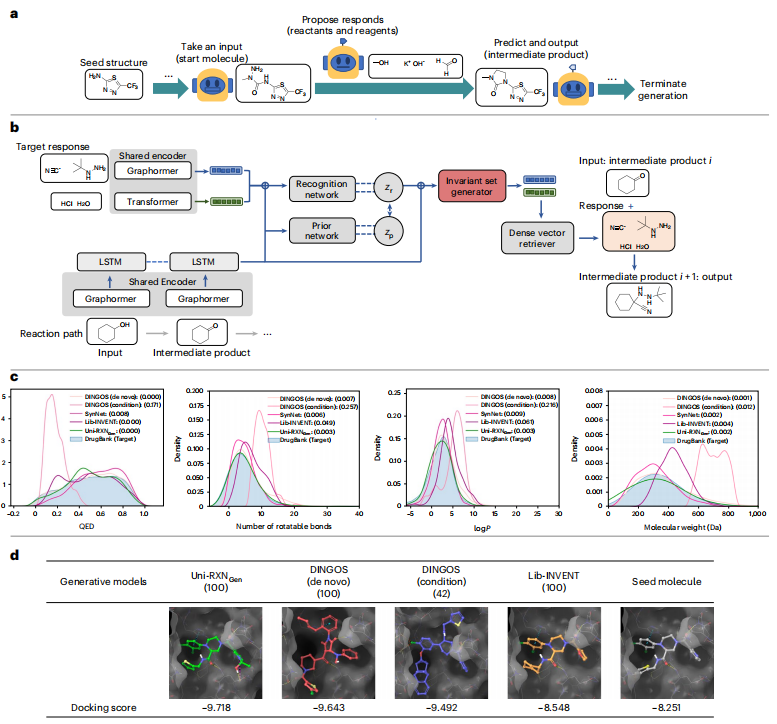
图示:Uni-RXNGen 流程与性能。(来源:论文)
与分子对接等虚拟筛选方法相结合,该生成模型可以实现高效的构效关系研究。该模型生成的巨大的可合成类药物化学空间可以提高药物再利用或命中分子搜索的真阳性率。
























