设计荧光分子需要考虑多种相互关联的分子特性,而不是与分子结构直接相关的特性,例如分子的光吸收。
在这项研究中,RIKEN 高级智能项目研究中心和东京大学等机构的研究人员合作,使用从头分子生成器(DNMG)与量子化学计算(QC)相结合来开发荧光分子,这些分子在各个学科中都引起了广泛关注。使用大规模并行计算(1024 核,5 天),DNMG 产生了 3643 个候选分子。
研究人员选择了一个未报道的分子和七个报道的分子并合成了它们。光致发光光谱测量表明,DNMG 可以以 75% 的准确度(n = 6/8)成功设计荧光分子,并产生一种未报告的分子,该分子发出肉眼可检测到的荧光。
该研究以「De novo creation of a naked eye–detectable fluorescent molecule based on quantum chemical computation and machine learning」为题,于 2022 年 3 月 9 日发布在《Science Advances》。

简介
荧光化合物作为可见光发射体在多个学科的应用中非常重要,包括有机发光二极管、传感器和生物成像。尽管已经为这些和其他应用开发了许多荧光分子,但不断需要新的分子来解决当前材料在功能、可持续性和低成本方面的缺点。即使是化学结构的细微变化也可能导致重大改进。
荧光是一种受量子力学支配的光化学性质。然而,尽管荧光研究的历史悠久,但没有明确的指导方针来制造荧光分子,就像制造吸光分子一样。
分子荧光发射的简化物理化学机制如下图所示。最初,研究人员认为分子处于单线态(S0)状态;在 S0 最小值时,它吸收光并转变为单线态第一激发态(S1)。S1 激发分子在 S1 状态下弛豫到最小值并回到 S0 状态,将 S1 和 S0 状态之间的能量差作为光(荧光)发射。激发的分子应该在 S1 状态下移动到最小值,以便在不失活的情况下发光。
一些因素,包括与氧分子的反应、分子碰撞、分子内/分子间电子转移和聚集,可能会在分子在激发状态下运动时使其失活;这导致很难将荧光与分子结构相关联。因此,自动化荧光分子设计将是有帮助的。
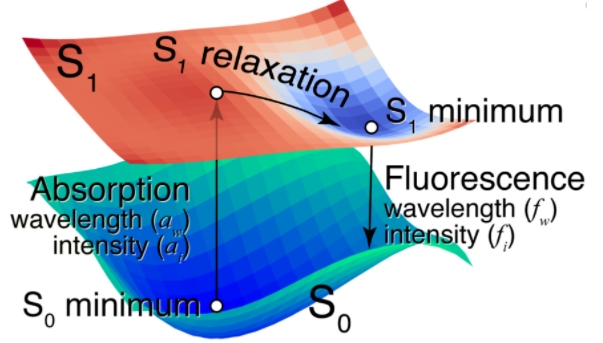
图示:荧光分子的单线态基(S0)态和单线态第一激发(S1)态的 PES 示意图。(来源:论文)
近期,基于机器学习(ML)的从头分子生成器(DNMG)已被开发用于设计具有简单和可预测值的分子,例如分配系数(logP)的对数,可以从分子的组成部分估计。
将 DNMG 与经典模拟相结合,成功地生成了具有改进的多功能性和实用性的分子。例如,DNMG 和对接模拟的结合可用于设计生物活性分子;这是通过有机合成研究的。结合分子动力学或预测模型,DNMG 还可以指导功能聚合物的合成。
在之前的研究中,研究人员将量子化学计算(QC)与 DNMG(称为 ChemTS)相结合,它(原则上)可以从头设计以量子力学(QM)特性为特征的功能分子。因此,ChemTS 与 QC 相结合被应用于设计可以吸收具有所需波长的光的分子。在 86 个设计和生成的分子中,选择了 6 个未包含在训练数据集中的已知分子进行紫外-可见 (UV-vis) 吸收测量。结果与发生器的目标波长一致。
此外,DNMG 增加了发现新分子的可能性,因为与传统的高通量 QM 和使用 ML 模型筛选相比,DNMG 的搜索区域在数据集中不受限制。研究人员还使用 QC 对 ChemTS 产生的分子进行了官能团富集分析,以最大化电子增益能量,并发现了驻极体文献中未包含的重要官能团。
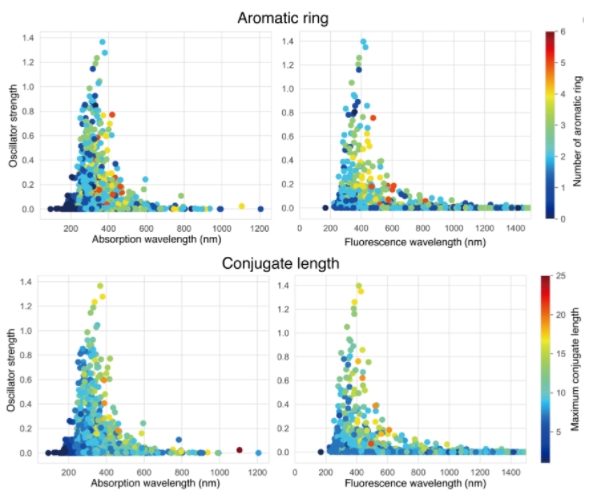
图示:在 B3LYP/3-21G* 水平上,生成分子的 S1 状态吸收和荧光的 OS 分布曲线。(来源:论文)
虽然相对简单的特性,如光吸收和电子增益能量,可以直接与分子结构相关,但复杂的现象,如仅由特定分子表现出的荧光,难度要大得多。
在荧光的情况下,有必要考虑多种性质,这些性质错综复杂地交织在一起。这使得很难为分子结构的设计建立直观的指导方针。为了设计实用的化合物,控制目标分子特性的复杂机制必须适当地数字化以用于 DNMG。此外,必须考虑计算成本随着探索化学空间的机制复杂性的增加而增加。
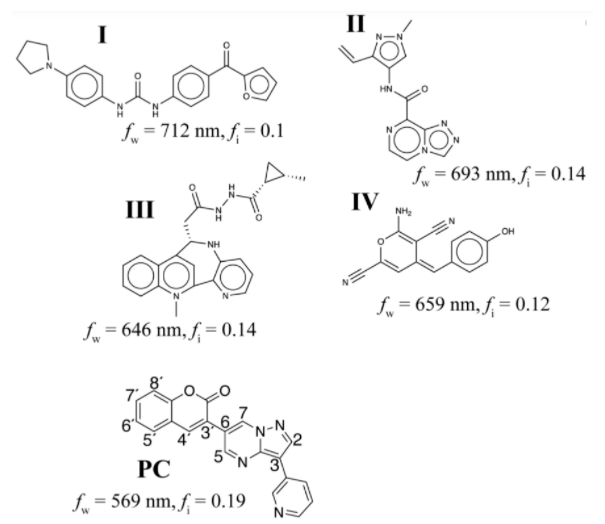
图示:使用 ChemTS 设计的未报告的荧光分子。(来源:论文)
在这项研究中,该团队设计了具有大规模并行化 ChemTS 版本的荧光分子。该程序包使用 QC 将荧光机制的最低要求数字化。有几个基于电子结构理论的用户友好软件包可用于分子和材料的 QC。
为了平衡可靠性与计算成本,研究人员使用密度泛函理论 (DFT) (29) 来评估势能面 (PES)。为了解决广泛探索化学空间的计算成本,用基于虚拟损失的概念对 ChemTS 进行大规模并行化;使用 1024 个核,生成了 3643 个分子。
为了验证,他们合成了一种未报告的化合物和几种已报告的化合物。六种化合物,包括一种未报道的化合物,如预期的那样发出荧光。未报道的分子可通过市售试剂之间的偶联合成,具有意想不到的特性,尽管它由常见的片段[香豆素、吡啶和吡唑并嘧啶]组成。这表明大规模并行 DNMG 有可能引发分子设计的范式转变。
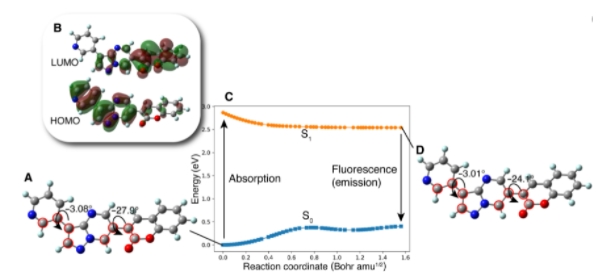
图示:PC的光致过程。(来源:论文)
讨论
几十年来,QC 在化学和材料科学中发挥了重要作用。在此期间,计算机辅助分子设计已被用于药物发现。然而,QC 只专注于对实验结果的分析和推测;很少考虑诸如预测各种现象和设计材料之类的创造性工作。
ML 算法最近在化学和材料科学中的应用,代表了计算机辅助化学和材料科学的一个积极转折点。为了自动设计对有机电子有用的分子,将 QC 与 DNMG 相结合至关重要,因为在此类应用中,量子力学不容忽视。然而,基于 QC 的 DNMG 的价值必须先得到证明,然后才能在实践中采用。
在这项研究中,研究人员使用 DNMG 来创建具有目前无法轻易预测的特性的分子:荧光。他们使用 DFT 设计了荧光化合物,这是一种固有的量子力学方法。尽管众所周知,分子受量子力学规则的支配,但仅凭 QC 很难从头创建一个新分子。尽管荧光分子具有简单的 PES,但它们很难从第一原理设计,因为它们的多样性使得荧光与分子结构的关联变得非常困难。
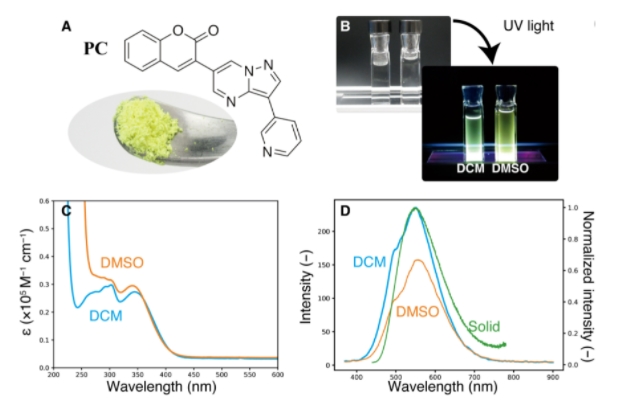
图示:PC的光化学性质。(来源:论文)
然而,生成器处理了这种多样性,并成功地从头开始设计了荧光分子。基于 QC 的大量从头计算需要大量并行计算(1024 核,5 天);尽管如此,生成器还是成功地产生了 3643 个候选分子。发生器产生吸收长波长光的分子,以类似于专业人士的方式控制分子的共轭长度;然而,它无法找到分子的荧光波长/强度与共轭长度/芳环数之间的明确相关性。这表明从头设计荧光分子的困难。
研究人员根据可合成性和可见荧光标准,选择了七种已知化合物进行验证和一种候选化合物进行进一步研究。实验验证表明,DNMG 成功设计了 75%(八分之六)的荧光化合物。PC 的荧光(肉眼可见)证明了 DNMG 的创新潜力。
在设计 PC 时,生成器在香豆素中引入了一个不熟悉的基团吡唑并嘧啶;这种连接引起了高空间排斥,但仍导致 OS 增加。
普通化学家很难想出一种通过增加片段之间的空间排斥来增强荧光及其强度的方法。这说明生成器是一种可以超越专业知识或直觉的工具。DNMG 有可能引发分子设计的范式转变。
虽然这项工作中合成的分子的荧光旨在通过肉眼检测,但通过消除对原子类型的限制并延长设计时间,将产生更多有趣的分子。
此外,通过包括分子的光诱导动力学,可以实现更好的分子设计。通过进一步开发 QC,可以设计出更复杂的功能分子。
因此,具有大规模并行计算的生成器将能够创建具有多样化和有趣功能的复杂分子,可能导致非常复杂的合成路线,这将增加最近开发的基于 ML 的规划逆合成路线的要求。
开源地址:
https://github.com/tsudalab/GaussianRunPack
https://github.com/tsudalab/FL_ChemTS