本文经AI新媒体量子位(公众号ID:QbitAI)授权转载,转载请联系出处。
喜大普奔!今天一波Nature、Science齐发文,可把学术圈的嗑盐人们高兴坏了。
一边是“AI界年度十大突破”AlphaFold2终于终于开源,登上Nature。
另一边Science又出报道:华盛顿大学竟然还搞出了一个比AlphaFold2更快更轻便的算法,只需要一个英伟达RTX2080 GPU,10分钟就能算出蛋白质结构!

要知道,当年AlphaFold2横空出世,那是真·沸腾了学术圈。
不仅谷歌CEO皮猜、马斯克、李飞飞等大V纷纷点赞,连马普所的演化生物研究所所长Andrei Lupas都直言:它会改变一切。
结构生物学家Petr Leiman感叹,我用价值一千万美元的电镜努力地解了好几年,Alphafold2竟然一下就算出来了。
更是有生物学网友表示绝望,感觉专业“前途渺茫”:

而今天这一波Nature、Science神仙打架,再次点燃话题度。
让学界狂热的Alphafold2
先说被顶刊争相报道的Alphafold2,它作为一个AI模型,为何引起各界狂热?
因为它一出来,就解决了生物学界最棘手的问题之一。这个问题于1972年被克里斯蒂安·安芬森提出,它的验证曾经困扰科学家50年:
给定一个氨基酸序列,理论上就能预测出蛋白质的3D结构。
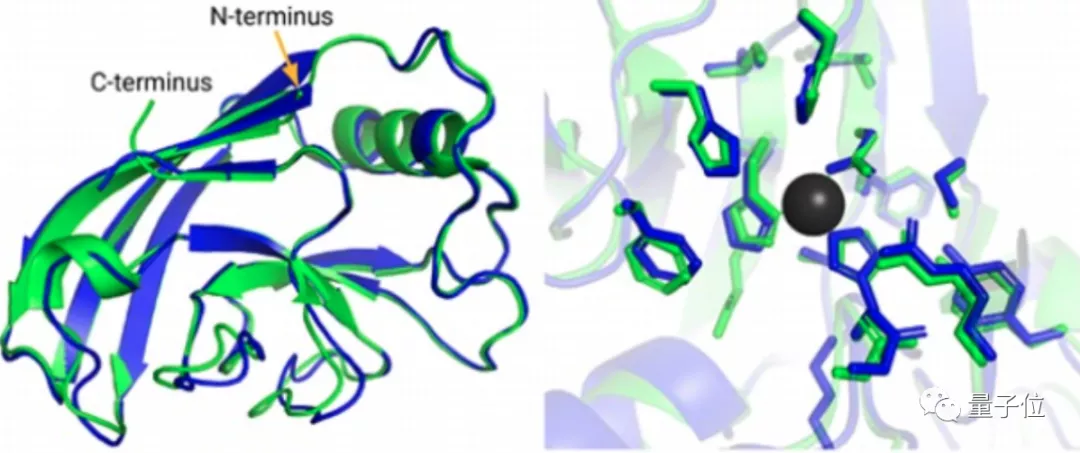
蛋白质由氨基酸序列组成,但真正决定蛋白质作用的,是它的3D结构,也就是氨基酸序列的折叠方式。
为了验证这个理论,科学家们尝试了各种手段,但在CASP14(蛋白质结构预测比赛)中,准确性也只达到40分左右(满分100)。
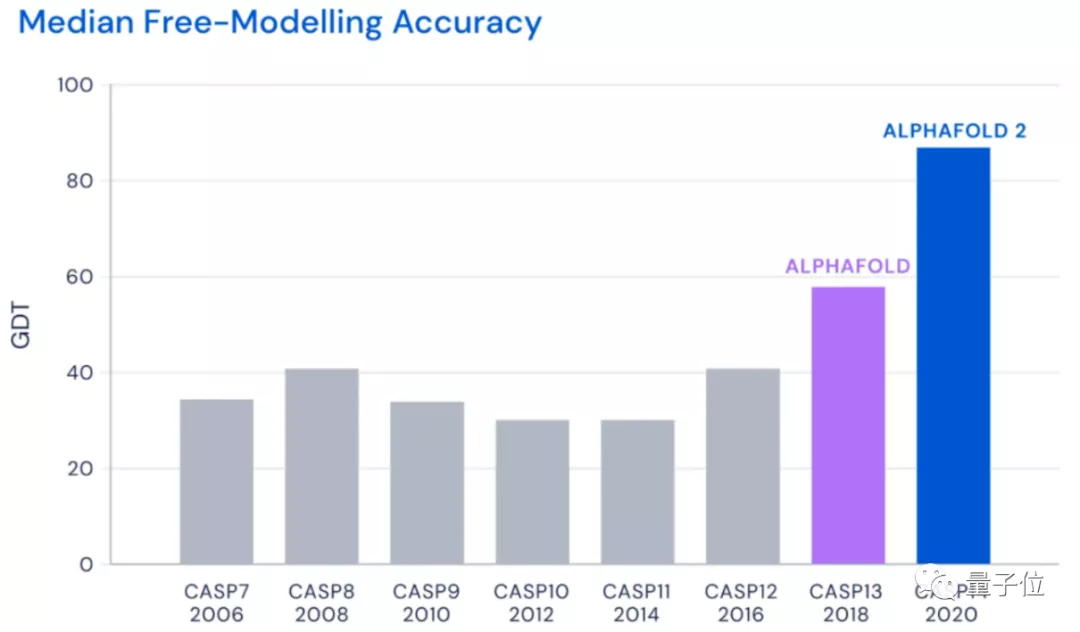
直到去年12月,Alphafold2出现,将这一准确性直接拔高到了92.4/100,和蛋白质真实结构之间只差一个原子的宽度,真正解决了蛋白质折叠的问题。
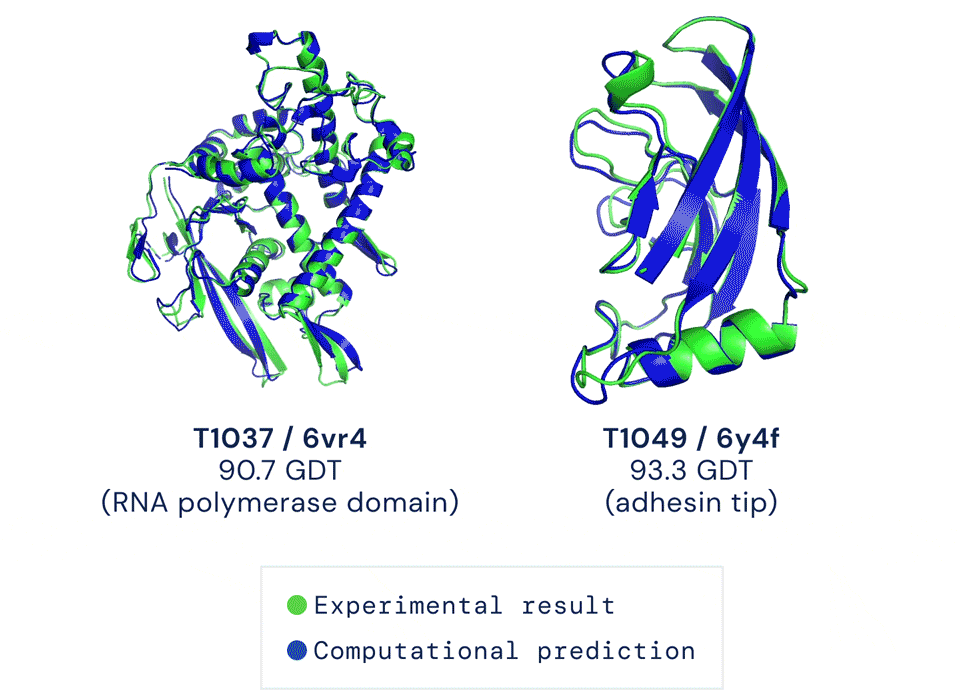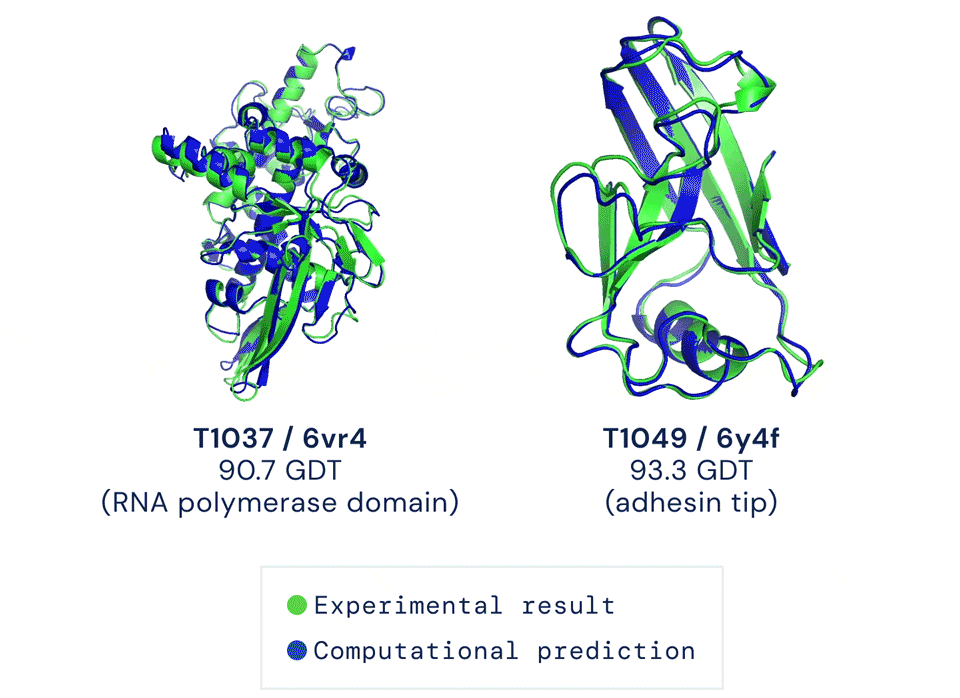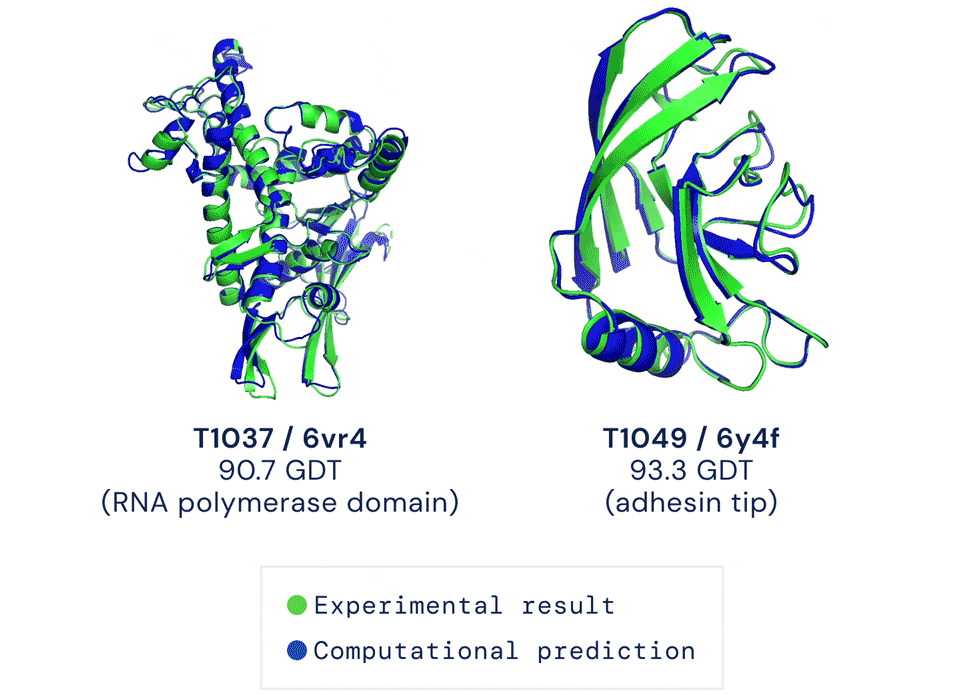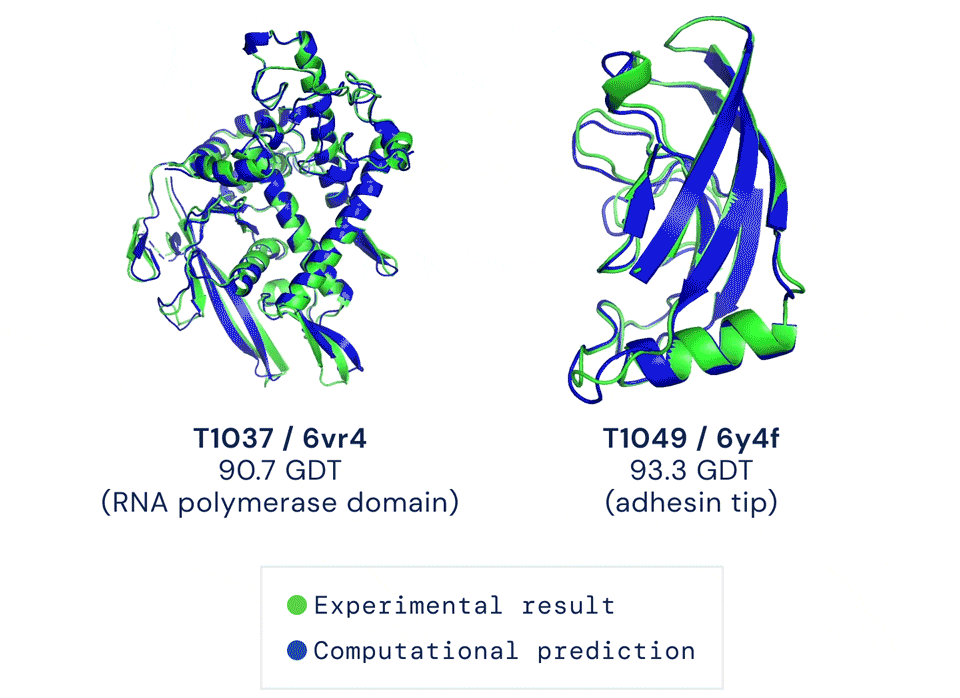
Alphafold2于当年入选Science年度十大突破,被称作结构生物学“革命性”的突破、蛋白质研究领域的里程碑。
它的出现,能更好地预判蛋白质与分子结合的概率,从而极大地加速新药研发的效率。
今天,Alphafold2的开源,又进一步在AI和生物学界激起了一大波浪。
谷歌CEO皮猜很高兴:

亦有生物学博士表示:未来已来!

来自UC伯克利AI实验室的博士Roshan Rao在看过后表示,这份代码看起来不仅容易使用,而且文档也非常完善。
现在,是时候借着这份开源算法,弄清Alphafold2的魔术是怎么变的了。
AlphaFold2详细信息公开
研究人员强调,这是一个完全不同于AlphaFold的新模型。
2018年的AlphaFold使用的神经网络是类似ResNet的残差卷积网络,到了AlphaFold2则借鉴了AI研究中最近新兴起的Transformer架构。
Transformer使用注意力机制兴起于NLP领域,用于处理一连串的文本序列。
而氨基酸序列正是和文本类似的数据结构,AlphaFold2利用多序列比对,把蛋白质的结构和生物信息整合到了深度学习算法中。
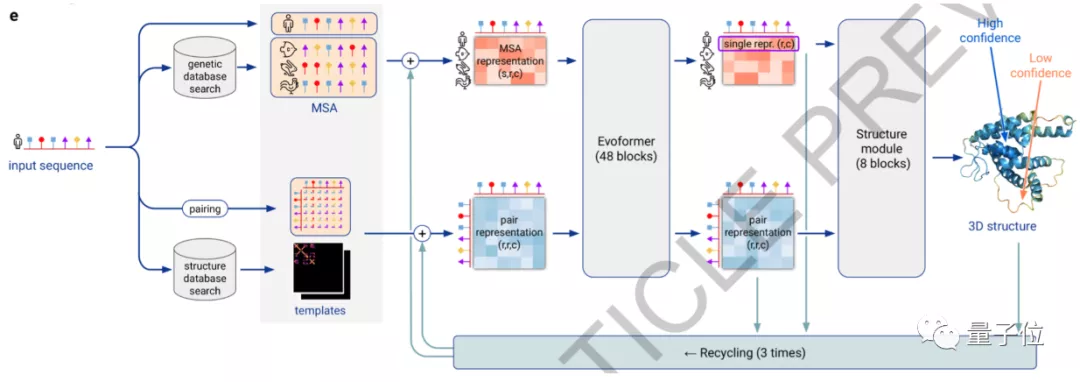
AlphaFold2用初始氨基酸序列与同源序列进行比对,直接预测蛋白质所有重原子的三维坐标。
从模型图中可以看到,输入初始氨基酸序列后,蛋白质的基因信息和结构信息会在数据库中进行比对。
多序列比对的目标是使参与比对的序列中有尽可能多的序列具有相同的碱基,这样可以推断出它们在结构和功能上的相似关系。
比对后的两组信息会组成一个48block的Evoformer块,然后得到较为相似的比对序列。
比对序列进一步组合8 blocks的结构模型,从而直接构建出蛋白质的3D结构。
最后两步过程还会进行3次循环,可以使预测更加准确。
△如何用三维坐标确定结构
还有更快、成本更低的算法?
AlphaFold2首次公布的时候并没有透露太多技术细节。
在华盛顿大学,同样致力于蛋白质领域的David Baker一度陷入失落:
如果有人已经解决了你正在研究的问题,但没有透露他们是如何解决的,你该如何继续研究?
不过他马上重整旗鼓,带领团队尝试能不能复现AlphaFold2的成功。
几个月后,Baker团队的成果不仅在准确度上和AlphaFold2不相上下,还在计算速度和算力需求上实现了超越。
就在AlphaFold2开源论文登上Nature的同一天,Baker团队的RoseTTAFold也登上Science。
RoseTTAFold只需要一块RTX2080显卡,就能在10分钟左右计算出400个氨基酸残基以内的蛋白质结构。
这样的速度,意味着什么?
那就是研究蛋白质的科学家不用再排队申请超算资源了,小型团队和个人研究者只需要一台普通的个人电脑就能轻松展开研究。
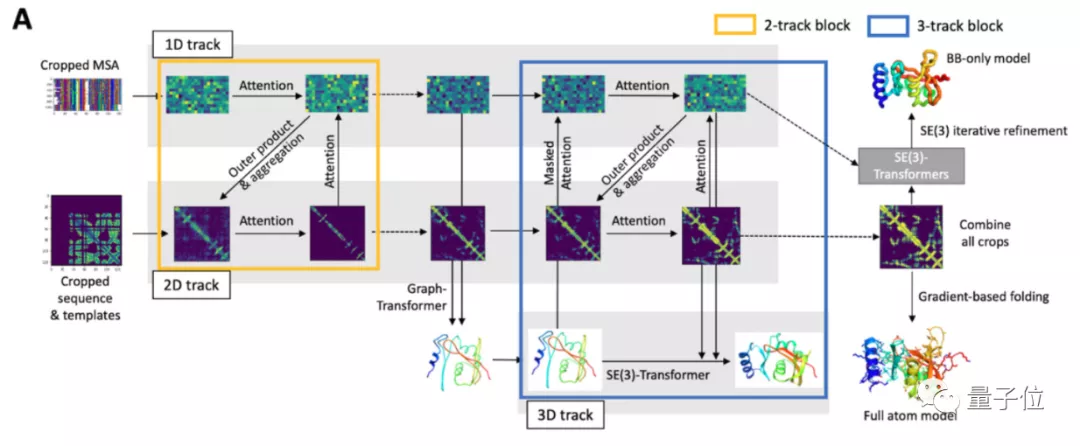
RoseTTAFold的秘诀在于采用了3轨注意力机制,分别关注蛋白质的一级结构、二级结构和三级结构。
再通过在三者之间加上多处连接,使整个神经网络能够同时学习3个维度层次的信息。
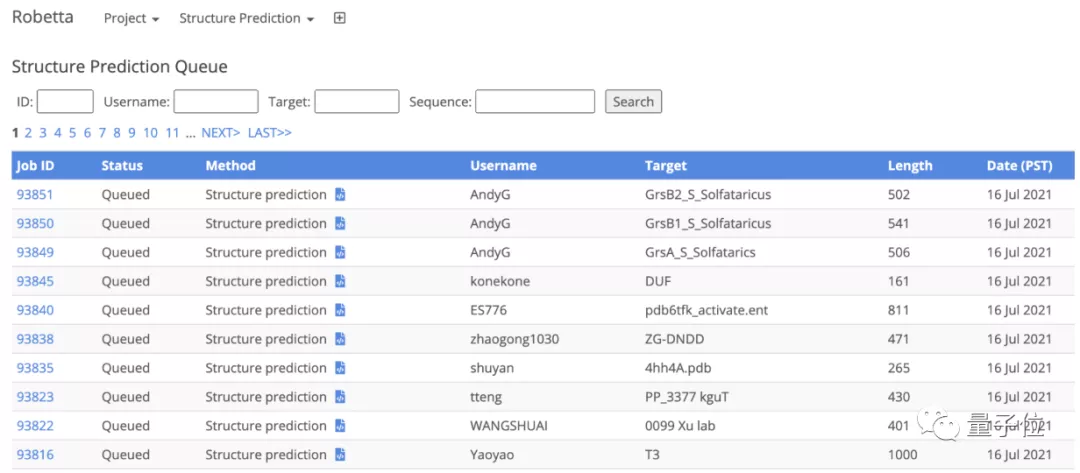
考虑到现在市场上显卡不太好买,Baker团队还贴心的搭建了公共服务器,任何人都可以提交蛋白质序列并预测结构。
自服务器建立以来,已经处理了来自全世界研究者提交的几千个蛋白质序列。
这还没完,团队发现如果同时输入多个氨基酸序列,RoseTTAFold还可以预测出蛋白质复合体的结构模型。
对于多个蛋白质组成的复合体,RoseTTAFold的实验结果是在24GB显存的英伟达Titan RTX上计算30分钟左右。
现在整个网络是用单个氨基酸序列训练的,团队下一步计划用多序列重新训练,在蛋白质复合体结构预测上还可能有提升空间。
正如Baker所说:
我们的成果可以帮助整个科学界,为生物学研究加速。
Alphafold2开源地址:
https://github.com/deepmind/alphafold
RoseTTAFold开源地址:
https://github.com/RosettaCommons/RoseTTAFold
相关论文:
Alphafold2:https://www.nature.com/articles/s41586-021-03819-2
RoseTTAFold:https://science.sciencemag.org/content/early/2021/07/14/science.abj8754