本文转载自公众号“读芯术”(ID:AI_Discovery)
2020年,在几何和图形机器学习论文中表现突出的,当属生物化学、药物设计和结构生物学。这可能是第一次,我们终于发现这些机器学习方法对基础科学的影响。本文中,我将重点介绍三篇论文,这三篇论文是过去一年内我感触最深的论文(笔者是其中一篇论文的共同作者)。
几何机器学习方法曾被刊登在《细胞》和《自然方法学》杂志2020年2月刊的封面上。
第一篇论文:
J. M. Stokes et al., A deep learning approach to antibiotic discovery (2020) Cell 180(4):688–702.
关于什么?基于图形神经网络研发抗菌药物的深度学习操作流程。
如何操作?经训练的图神经网络用于预测大肠杆菌在多于2000个已知抗菌活性分子(包括批准抗生素、动植物提取物)数据集上的生长抑制。这种预测只是基于分子图,并不依赖于任何其他辅助信息,如药物作用机制等。
训练模型被送到药物再利用中心,经调查研究,模型中含有约6000种药物分子,前100种分子被选作试验测试对象。令人吃惊的是,一种实验抗糖尿药物halicin(海利霉素)具备有效的抗菌效果,能够消灭实验小鼠体内的多种抗药病菌。
显然,图神经网络具备良好普适性,因为halicin分子不同于传统抗生素。但是在这篇论文中,还并不清楚这种预测能力是否可以归结为预测一种抗菌作用的简单模式(细胞膜去极化)。
此外,研究人员还对ZINC15数据库中超过1亿个分子结构进行实验筛选,ZINC15数据库是专门为虚拟筛选而准备的商业可用化合物数据库,通常为药物设计者所用。在挑选的化合物中,物理试验鉴定出8种具有抗菌活性,其中2种对多种病原体均有较强的活性。
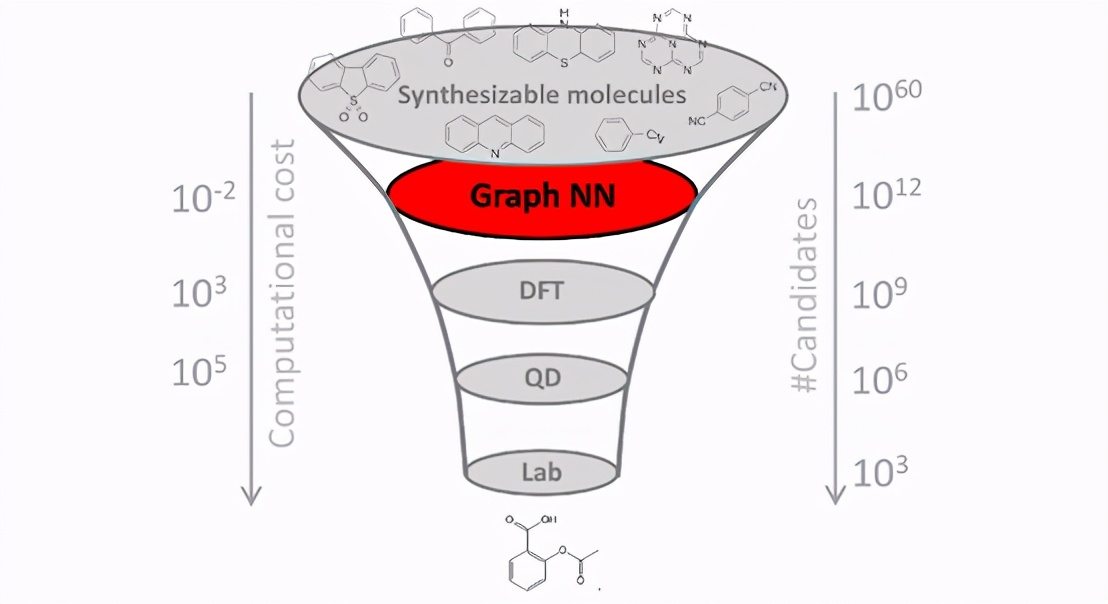
研发药物的一大挑战是,搜索空间很大,但是只有少数分子能够在实验室中测试。应用于分子图的图神经网络可用来预测分子属性,从而对所选药物进行虚拟筛查。
为何重要?巨大的搜索空间是研发药物的一大挑战,据估计,其中至少包含1060个分子。只有很少一部分分子能够在实验室中测试,挑选较有可能性的分子至关重要。通过计算方法完成挑选的过程称为“虚拟筛选”。
过去,机器学习方法经常用于分子的虚拟筛选,更广泛来说,协助不同阶段的药物研发,这是第一次在完全没有任何人类预假设的情况下,从零开始识别一种全新抗生素的过程。
大多数经由电脑模拟的、基于机器学习的药物研发论文结果都仅由计算机预测,但是斯托克斯等人的论文与之不同,他们的论文不仅鉴别有潜力的药物分子,而且在实验动物上广泛验证它们在活体内的活性。
虽然在原则上这种方法只可以用于寻找治疗癌症等疾病的方法,但是对于抗生素的关注非常及时:滥用抗生素导致抗药微生物形成,成为威胁全人类健康的梦魇,而且可能出现高传染性的细菌感染,现有药物无法治疗,这个现象肯定会出现,只是时间问题而已。
更多相关内容:《量子杂志》热搜文章和吉姆·柯林斯(Jim·Collins)2020年TED演讲视频(柯林斯实验室是本年TED“无畏项目”之一,我们的CETI项目也属于“无畏项目”)。
第二篇论文:
Jumper et al., High accuracy protein structure predictionusing deep learning (2020) a.k.a. AlphaFold 2.0 (尚未提供全文)
关于什么?根据氨基酸序列预测蛋白质的3D结构,这是生物信息学领域众所周知的一个难题。
如何操作?AlphaFold 2.0是一个“基于注意力的神经网络”(类似于变压器结构),对蛋白质数据库中17万种蛋白质结构和未知结构蛋白质序列进行端对端训练。但是DeepMind尚未公布算法细节,我们只能假设它是如何工作的。
在这篇文章中,蛋白质被建模为空间联系图,神经网络“解释该图的结构,同时对正在构建的隐图进行推理”。这听起来很像具备潜在图学习功能的图神经网络,只不过其中可能包含更多附加细节和细微差异,因为这种方法也使用进化序列信息,所以我将其归类为“几何机器学习”。
据报道,训练的计算复杂度很高(相当于数年的GPU时间),而对结构的预测不过是“几天的事”。
魔蛇玩具形象地展现了蛋白质折叠,在蛋白质折叠过程中氨基酸的一维序列折叠成复杂的3D形状,赋予蛋白质功能
为何重要?蛋白质可以说是最重要的生物分子,经常被称为“生命分子”,我们还未见过任何不以蛋白质为基础的生命形式。蛋白质在DNA内编码,在体内具备各种功能,包括抵抗病原体(抗生素)、形成皮肤结构(胶原蛋白)、输送氧气到细胞(血红蛋白)、催化化学反应(酶)及信号传递(许多激素是蛋白质)。
从化学角度来讲,蛋白质是生物聚合物或者由氨基酸组成的链,在静电作用下折叠成复杂的3D结构。正是这种结构赋予蛋白质功能,而且这种结构对理解蛋白质是如何工作以及做什么是非常必要的。蛋白质一般是药物治疗的靶点(药物是设计成与靶点相结合的小分子),所以制药业极为关注该方面研究。
现代技术可以对蛋白质进行排列(即形成氨基酸串),而且成本较低、技术可靠,不过获取3D结构主要还是依赖于传统的结晶技术,尽管结晶技术不稳定、耗时长、成本高。目前,已知序列的蛋白质大约有2亿种,已知结构的蛋白质至少有20万种蛋白质。
一直以来,人们认为氨基酸序列中包含了足够多预测蛋白质结构的信息,但是现在这个观点站不住脚了。蛋白质结构预测关键技术分析大赛(CASP)是类似于ImageNet的竞赛,自1994年开始举办,参赛者需要预测未知蛋白质的3D结构,这个大赛已成为生物信息实验室和制药公司的经典测试平台。
2018年,DeepMind的新技术AlphaFold在CASP大赛中脱颖而出,获得比赛胜利,震惊研究界。2020版AlphaFold 2.0效果更好,均方根误差仅1.6埃,按照结构生物学标准可以说是非常精确,远远超过其他竞争对手。这是蛋白质科学领域里的“ImageNet时刻”。
尽管在关键问题上取得了惊人的进展,但是媒体大肆炒作、用词随意,歪曲了AlphaFold的功能。特别是在药物设计应用上,结合部位通常需要达到亚埃精确度,但是这项技术尚未实现该功能。
更多相关内容:每个人都热切期待解释该算法的论文发表。莱克斯·弗里德曼(Lex Fridman)在YouTube视频中进行了很好的概括,穆罕默德·艾尔库雷希(Mohammed AlQuraishi)在博客中介绍了AlphaFold在2018年的影响。
第三篇论文:
P. Gainza et al., Deciphering interactionfingerprints from protein molecular surfaces using geometric deep learning (2020) Nature Methods 17(2):184–192.
关于什么?一个名为MaSIF的几何深度学习方法从蛋白质的3D结构预测蛋白质之间的相互作用。
如何操作?MaSIF将蛋白质模拟为一个离散成网格的分子界面,研究人员认为此种方式在处理相互作用时是有利的,因为它可以提取出内部的折叠结构。这个架构是基于MoNet发明的,MoNet是我的博士研究生费德里科·蒙奇发明的一个网状卷积神经网络,基于预先计算的小地测片中的化学和几何特点。
该网络使用蛋白质数据库中的几千个共晶蛋白质3D结构来进行训练,从而解决界面预测、配基分类和对接等各种问题,展现现代化的性能。MaSIF与其他方法最大的差异是,它不依赖于蛋白质的进化史。这在蛋白质全新设计中至关重要,尝试“从头”创造前所未有的全新蛋白质。
作为本篇论文的共同作者,我要强调的是预算分子界面和本地补丁的重要性,而且手工制作特性的依赖性是MaSIF的主要缺点之一。
在这一年里,我们彻底改造了结构,直接操作原子点云来输入,飞速计算分子界面(表现为点云),学习几何和化学特征,端到端可辨,运行速度快了几个数量级(后者是通过使用快速几何计算库KeOps实现的,是我的博士后珍·菲迪(Jean Feydy)发明的)。
虽然《自然方法》论文主要关注计算方法,但是随后EPFL的合作者获得了MaSIF设计的几种新型蛋白质结合剂的晶体结构,其与所计算结构高度吻合。
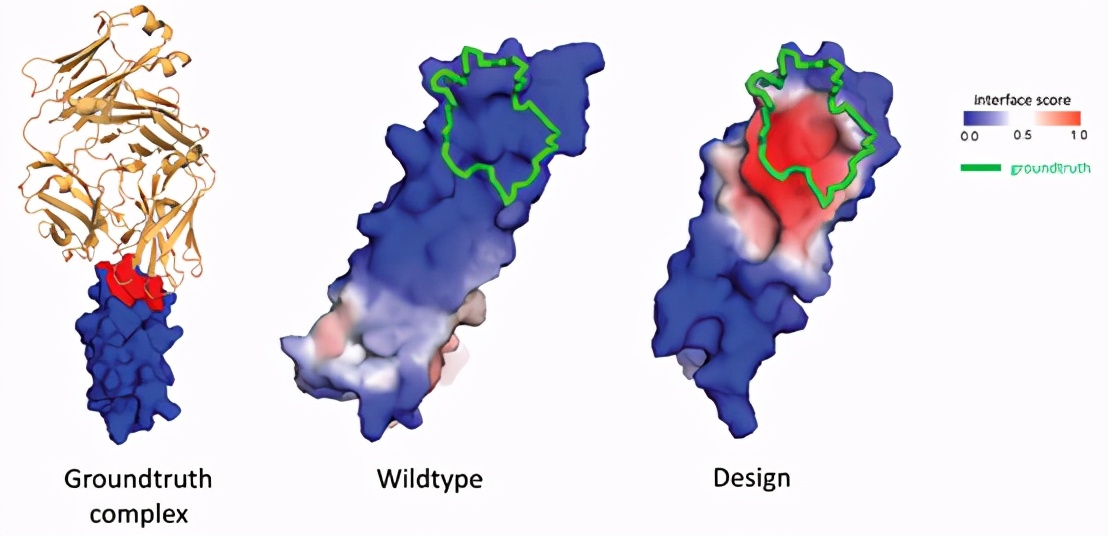
使用MaSIF预测蛋白质的结合位置。如图所示设计蛋白质(右)经过修饰,以改善与自然产生的“野生型”(中)靶点相结合。即使结合部位结构偏平,MaSIF也可以准确探测其位置。
为何重要?蛋白质与其他生物分子之间的相互作用是大多数生物活动中蛋白质发挥功能的基础。更好地理解蛋白质的作用原理对基础生物学和药物研发都非常重要,许多疾病与蛋白质间相互作用(PPI)有关,这种相互作用是理想的药物靶点。然而,这种相互作用通常包含“不可药物治疗”的扁平界面,因为他们与传统的小药分子靶向的口袋型结构大不相同。
MaSIF能够成功识别靶点的结合剂,是理性蛋白质设计的理想工具,开启了生物药物研究的各种应用,比如免疫抑制检查站癌症治疗,这种疗法以负责程序性细胞死亡的PD-1/PD-L1蛋白质复合体为靶体。